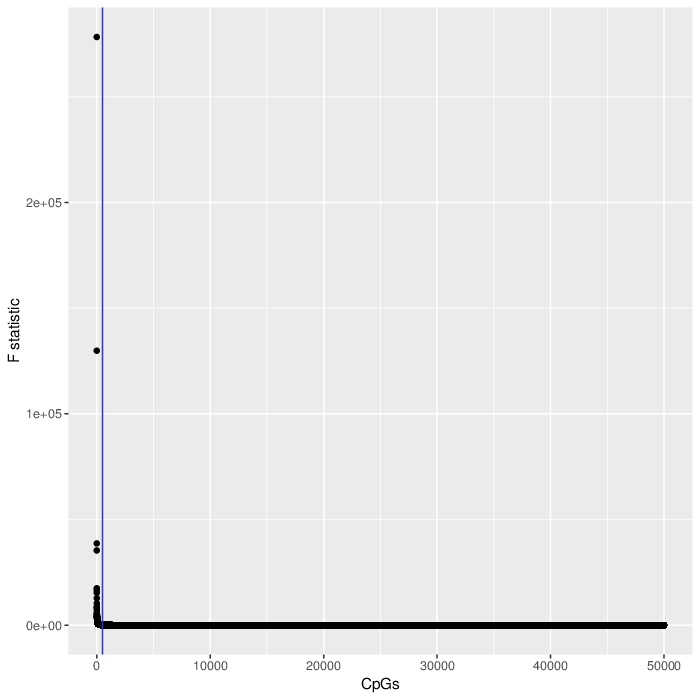
In the first step the reference methylomes were used to estimate the association of each CpG position to each of the cell types. The stength of association was measured using F-test. To decrease the computation load, only 50000 most variable CpGs were considered. Finally, only 500 CpGs with the lowest F-test p-value were used in the contribution estimation. The plot below visualizes the distribution of F statistic values for all tested CpGs. Note that selecting the most informative CpGs is equivalent to applying an F statistic cut-off of 83.6.
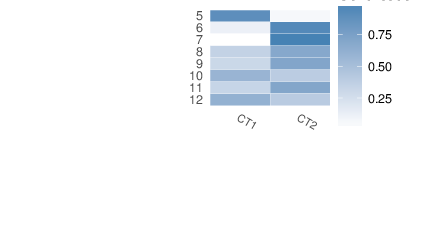
After the marker selection, a projection of the target data onto the space of the marker selection model coefficients yields contributions of each reference cell type to each measured DNA methylation profile. The resulting cell type contributions are available in a dedicated comma-separated file accompanying this report. These values are also displayed in the heatmap below. The contributions are constrained to be greater or equal to zero, and the per-sample sums are expected to be close to one, i.e. they are estimates of the cell type proportions. Per-sample totals much larger than one may indicate the problems with the procedure, e.g. bad correspondence of the target data to the reference methylomes, significant batch effects etc.