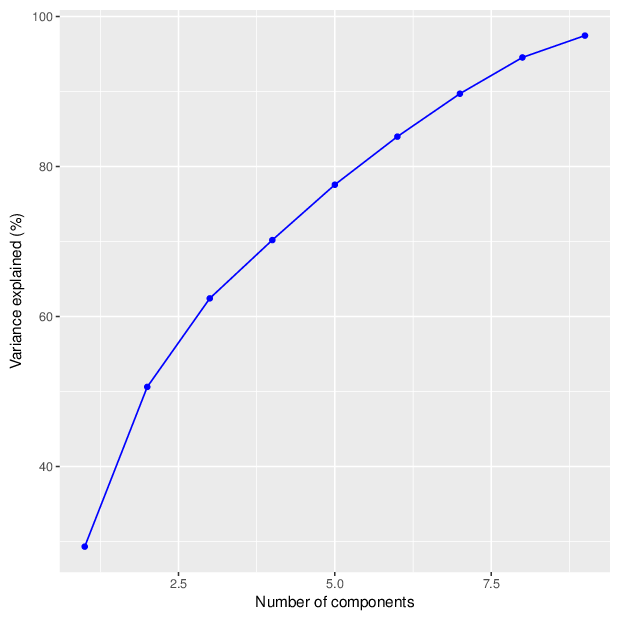
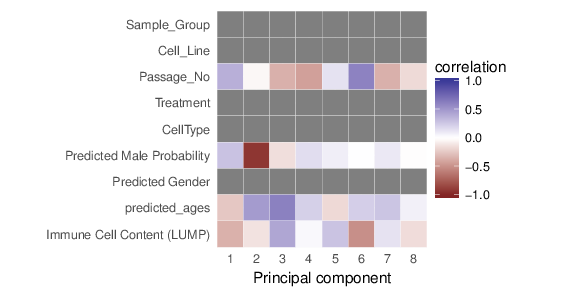
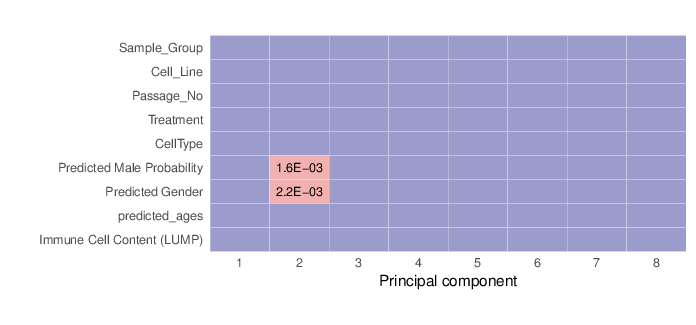
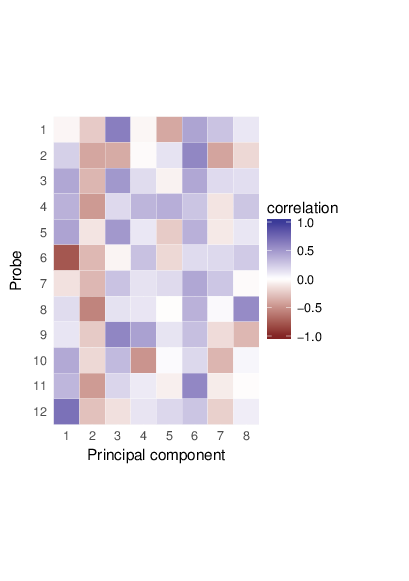
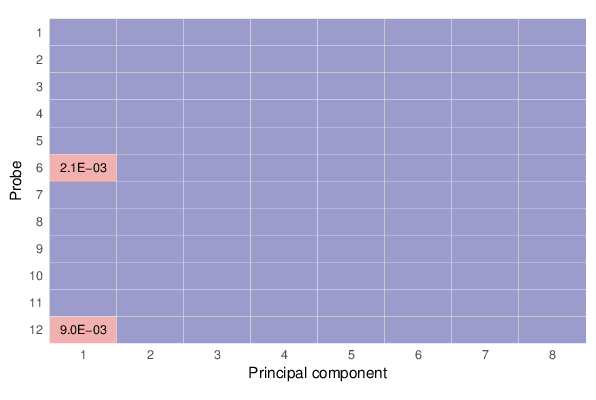
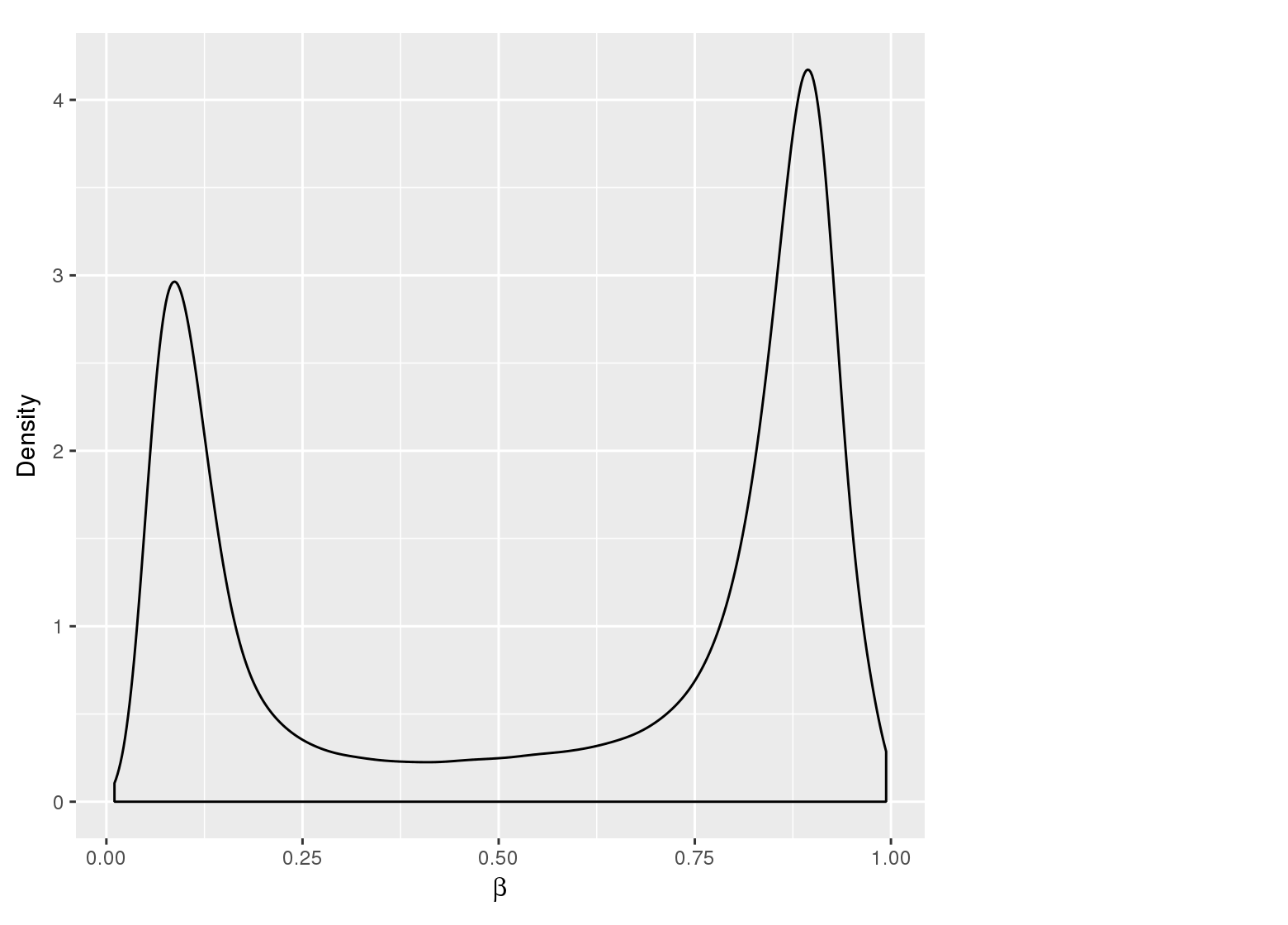
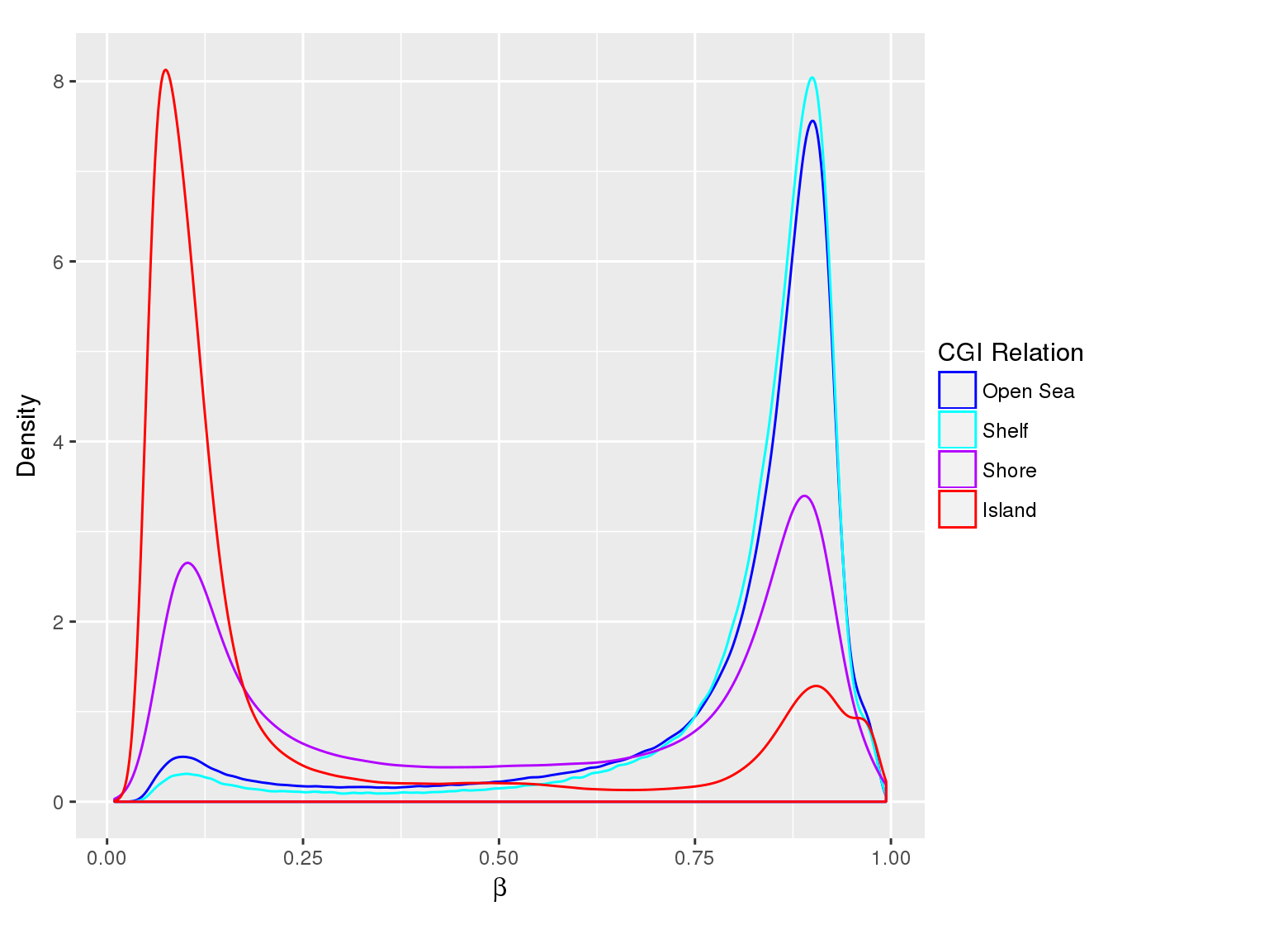
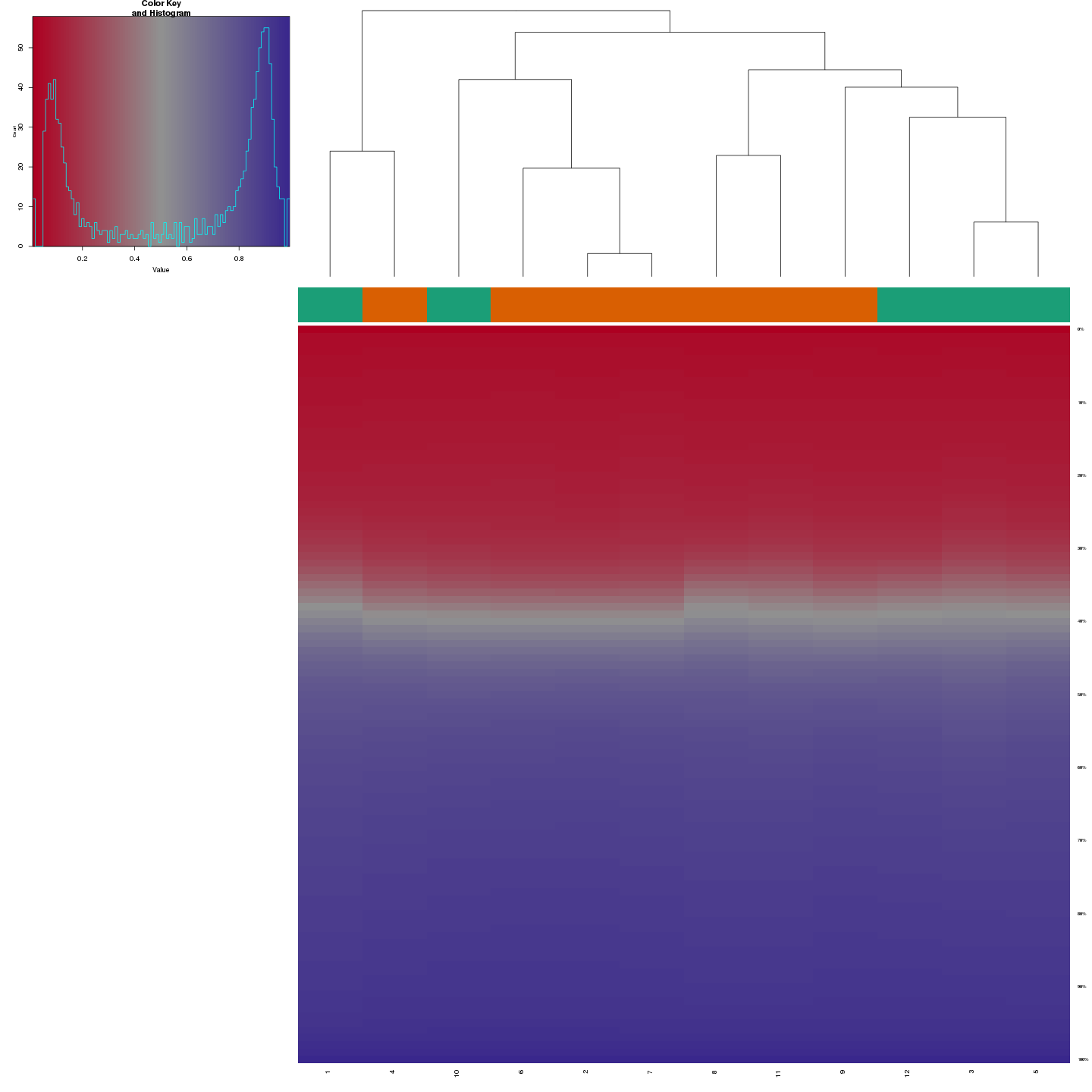
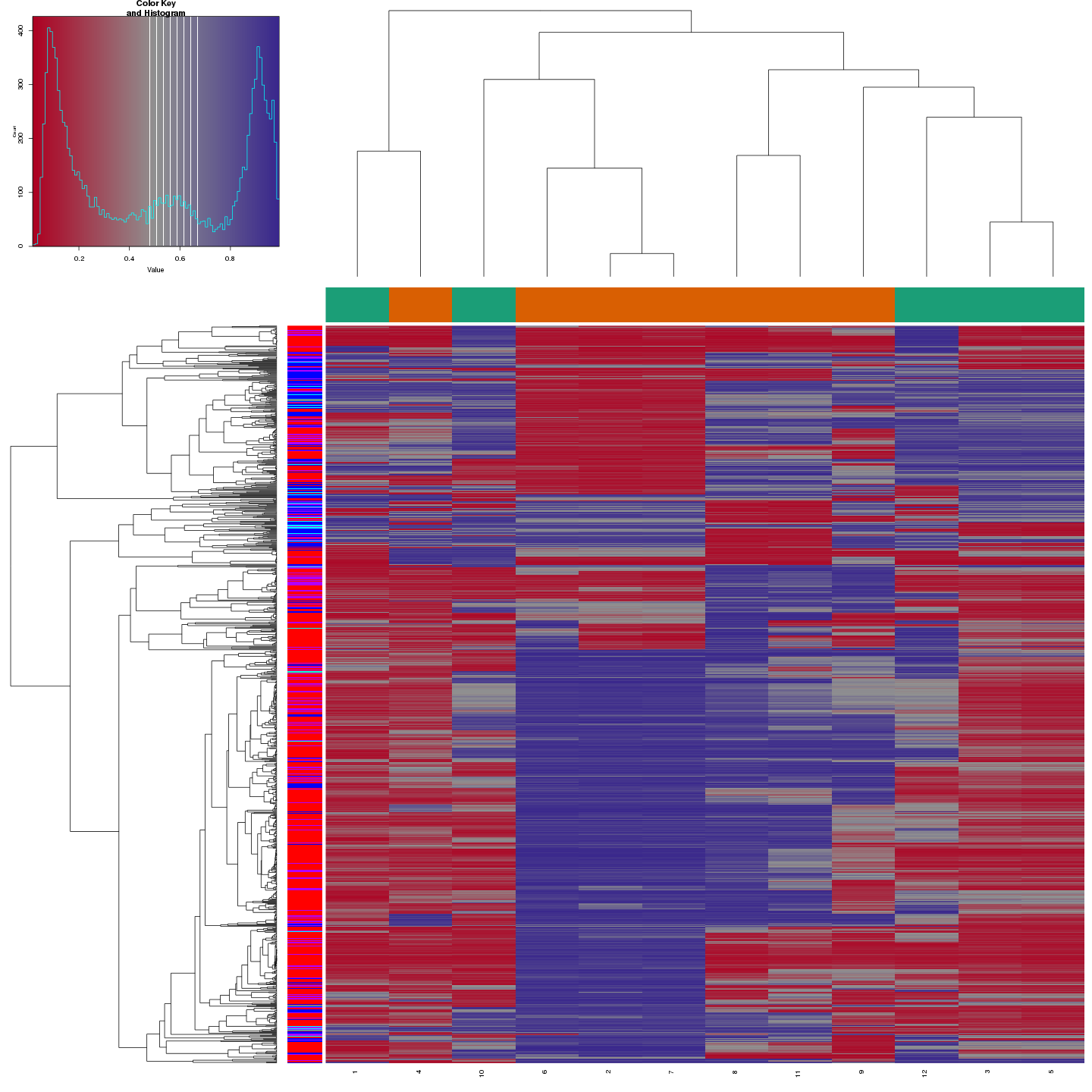
Methylation value distributions were assessed based on selected sample groups. This was done on probe and region levels. This section contains the generated density plots.
The plots below compare the distributions of methylation values in different sample groups, as defined by the traits listed above.
In a similar fashion, the plot below compares the distributions of beta values in different probe types.
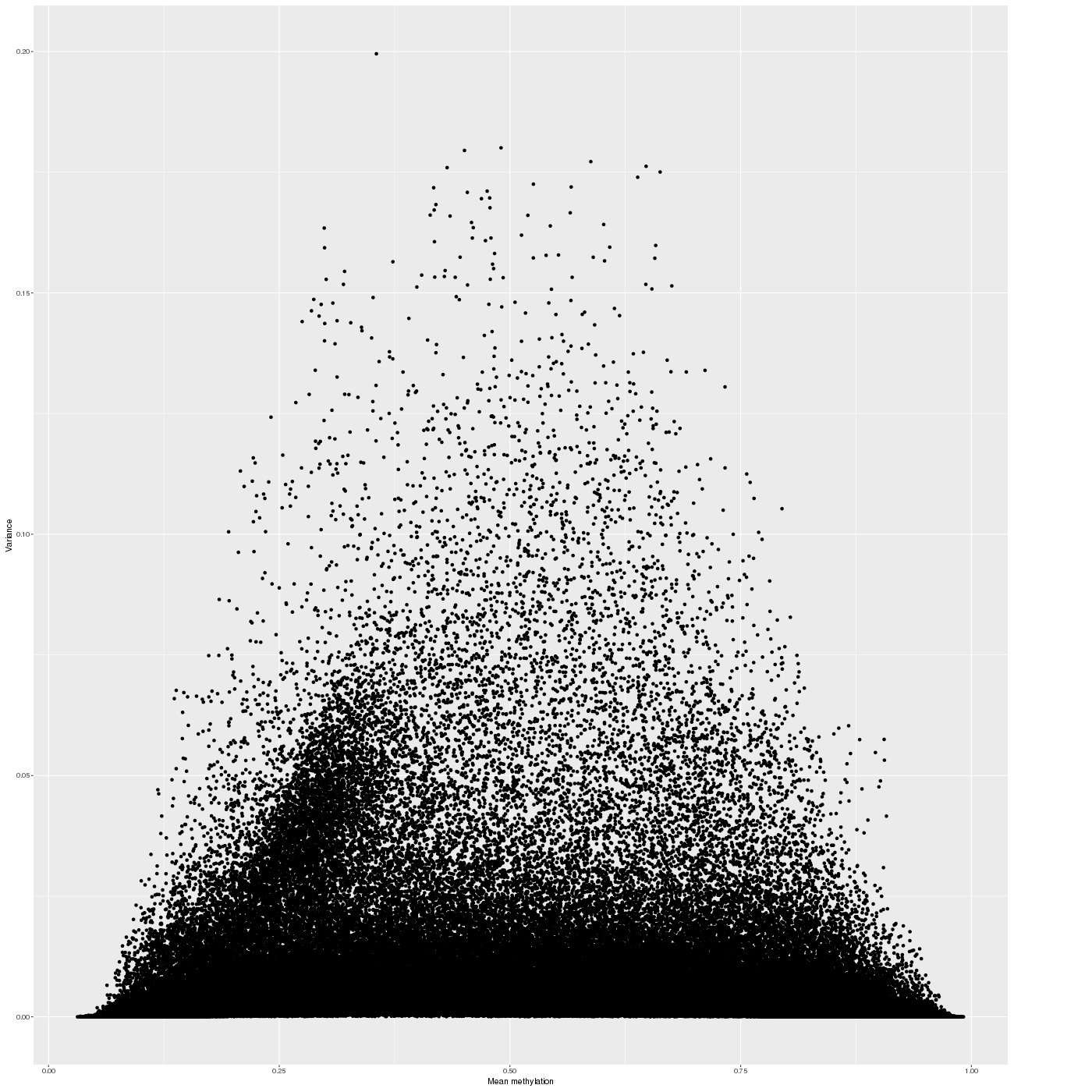
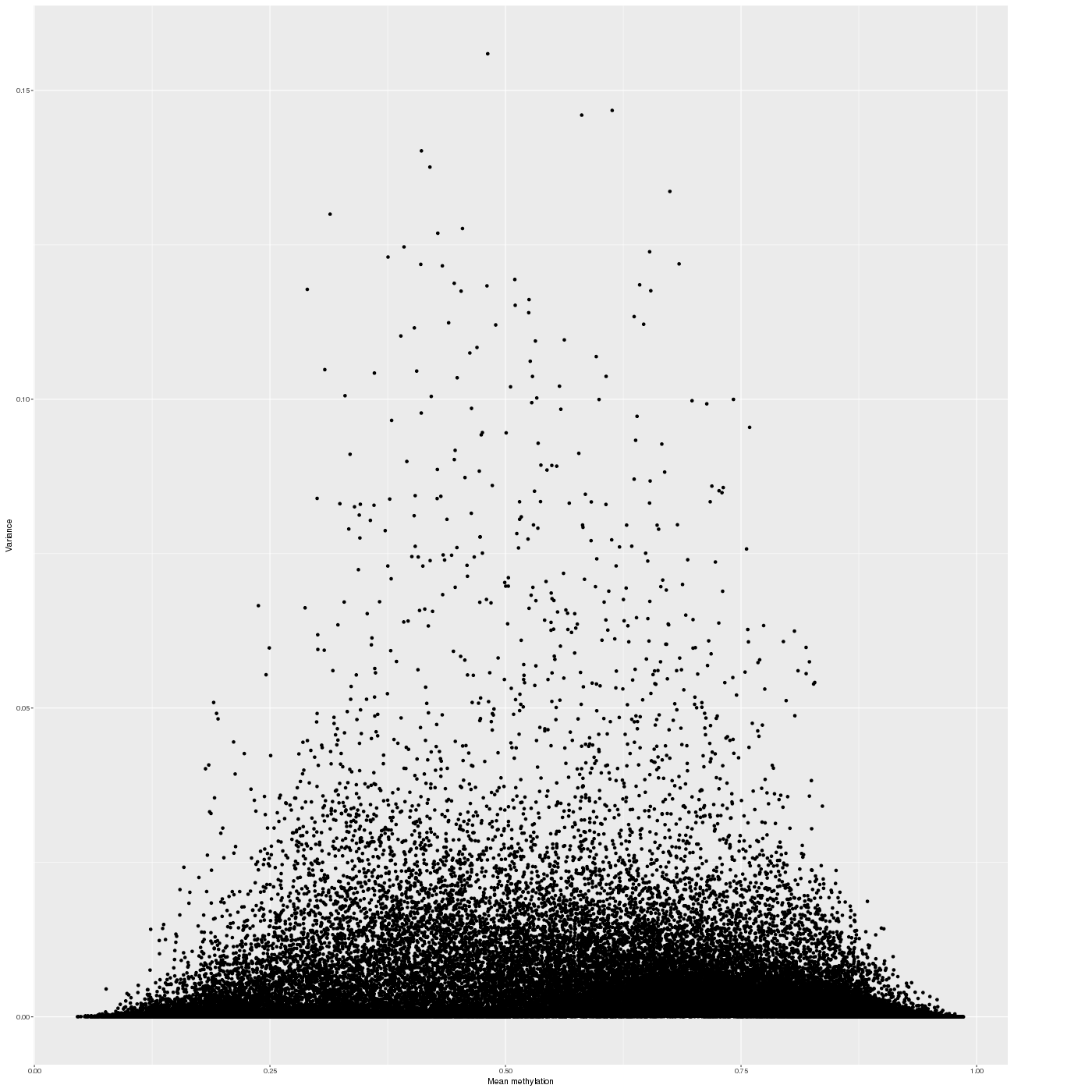
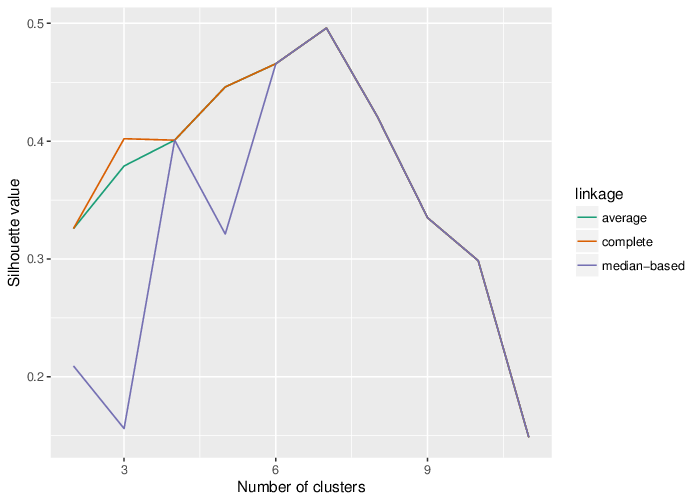
The variability of the methylation values is measured in two aspects: (1) intra-sample variance, that is, differences of methylation between genomic locations/regions within the same sample, and (2) inter-sample variance, i.e. variability in the methylation degree at a specific locus/region across a group of samples.
The following figure shows the relationship between average methylation and methylation variability of a probe.
In a complete analogy to the plots above, the figure below shows the relationship between average methylation and methylation variability of a genomic region.
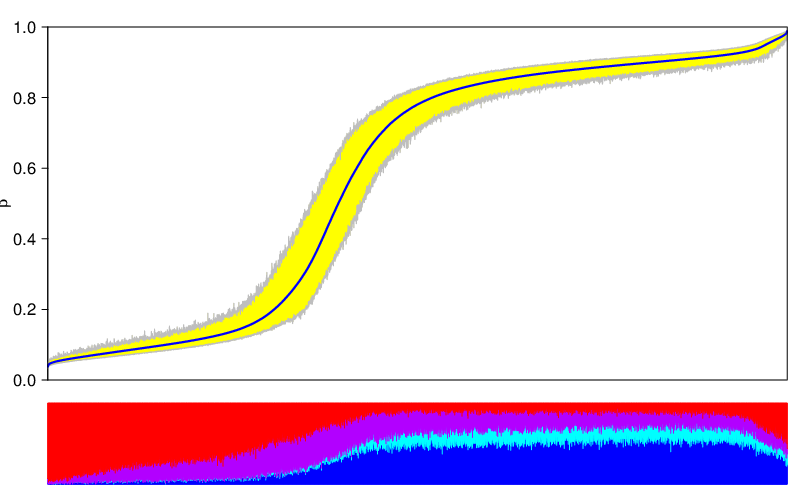
The figure below shows a methylation deviation plot for all samples in the dataset, as well as other sample groups inferred from the table of phenotypic information.
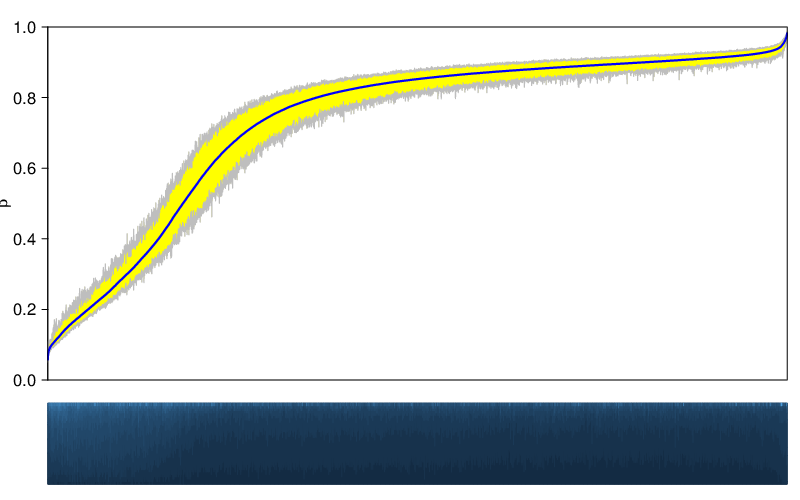
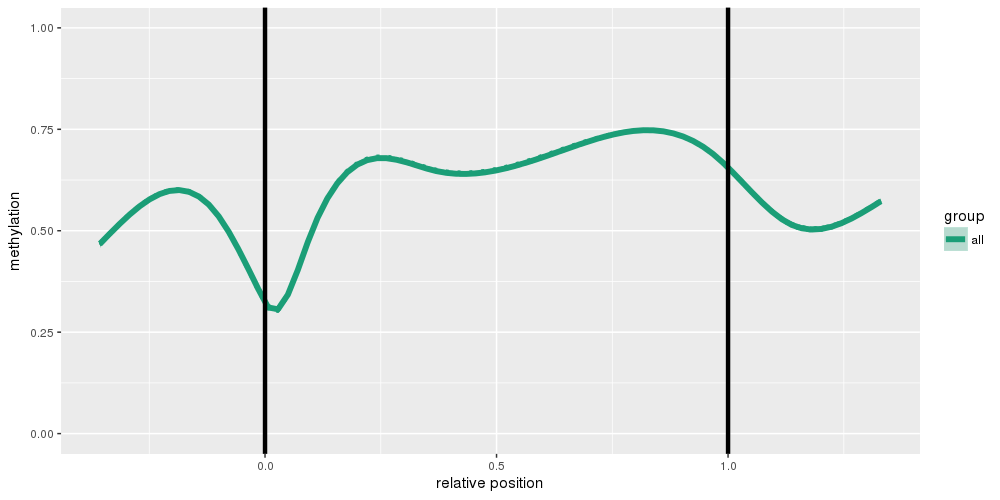
In a similar fashion, the figure below shows deviation plots on the region level.
The variability of a sample group is the span between 5th and 95th percentile of β values , averaged over all valid locations/regions. This amounts to a number between 0 and 1 and corresponds to the relative area of deviation in the plots presented above. The table below lists the variabilities of the studied sample groups.
| sites |
all samples |
|
12 |
0.0890 |
| sites |
hESC |
Sample_Group |
5 |
0.0665 |
| sites |
hiPSC |
Sample_Group |
7 |
0.0746 |
| sites |
KOSR |
Treatment |
2 |
0.0294 |
| sites |
TeSR |
Treatment |
2 |
0.0351 |
| sites |
CT1 |
CellType |
2 |
0.0360 |
| sites |
CT2 |
CellType |
2 |
0.0336 |
| sites |
female |
Predicted Gender |
6 |
0.0685 |
| sites |
male |
Predicted Gender |
6 |
0.0715 |
| tiling |
all samples |
|
12 |
0.0784 |
| tiling |
hESC |
Sample_Group |
5 |
0.0573 |
| tiling |
hiPSC |
Sample_Group |
7 |
0.0661 |
| tiling |
KOSR |
Treatment |
2 |
0.0239 |
| tiling |
TeSR |
Treatment |
2 |
0.0311 |
| tiling |
CT1 |
CellType |
2 |
0.0306 |
| tiling |
CT2 |
CellType |
2 |
0.0295 |
| tiling |
female |
Predicted Gender |
6 |
0.0625 |
| tiling |
male |
Predicted Gender |
6 |
0.0639 |
| genes |
all samples |
|
12 |
0.0610 |
| genes |
hESC |
Sample_Group |
5 |
0.0454 |
| genes |
hiPSC |
Sample_Group |
7 |
0.0510 |
| genes |
KOSR |
Treatment |
2 |
0.0206 |
| genes |
TeSR |
Treatment |
2 |
0.0248 |
| genes |
CT1 |
CellType |
2 |
0.0244 |
| genes |
CT2 |
CellType |
2 |
0.0226 |
| genes |
female |
Predicted Gender |
6 |
0.0461 |
| genes |
male |
Predicted Gender |
6 |
0.0471 |
| promoters |
all samples |
|
12 |
0.0693 |
| promoters |
hESC |
Sample_Group |
5 |
0.0523 |
| promoters |
hiPSC |
Sample_Group |
7 |
0.0578 |
| promoters |
KOSR |
Treatment |
2 |
0.0231 |
| promoters |
TeSR |
Treatment |
2 |
0.0281 |
| promoters |
CT1 |
CellType |
2 |
0.0281 |
| promoters |
CT2 |
CellType |
2 |
0.0255 |
| promoters |
female |
Predicted Gender |
6 |
0.0519 |
| promoters |
male |
Predicted Gender |
6 |
0.0533 |
| cpgislands |
all samples |
|
12 |
0.0781 |
| cpgislands |
hESC |
Sample_Group |
5 |
0.0581 |
| cpgislands |
hiPSC |
Sample_Group |
7 |
0.0652 |
| cpgislands |
KOSR |
Treatment |
2 |
0.0290 |
| cpgislands |
TeSR |
Treatment |
2 |
0.0317 |
| cpgislands |
CT1 |
CellType |
2 |
0.0307 |
| cpgislands |
CT2 |
CellType |
2 |
0.0285 |
| cpgislands |
female |
Predicted Gender |
6 |
0.0568 |
| cpgislands |
male |
Predicted Gender |
6 |
0.0589 |