This section contains quality control plots and statistics for the methylation data.
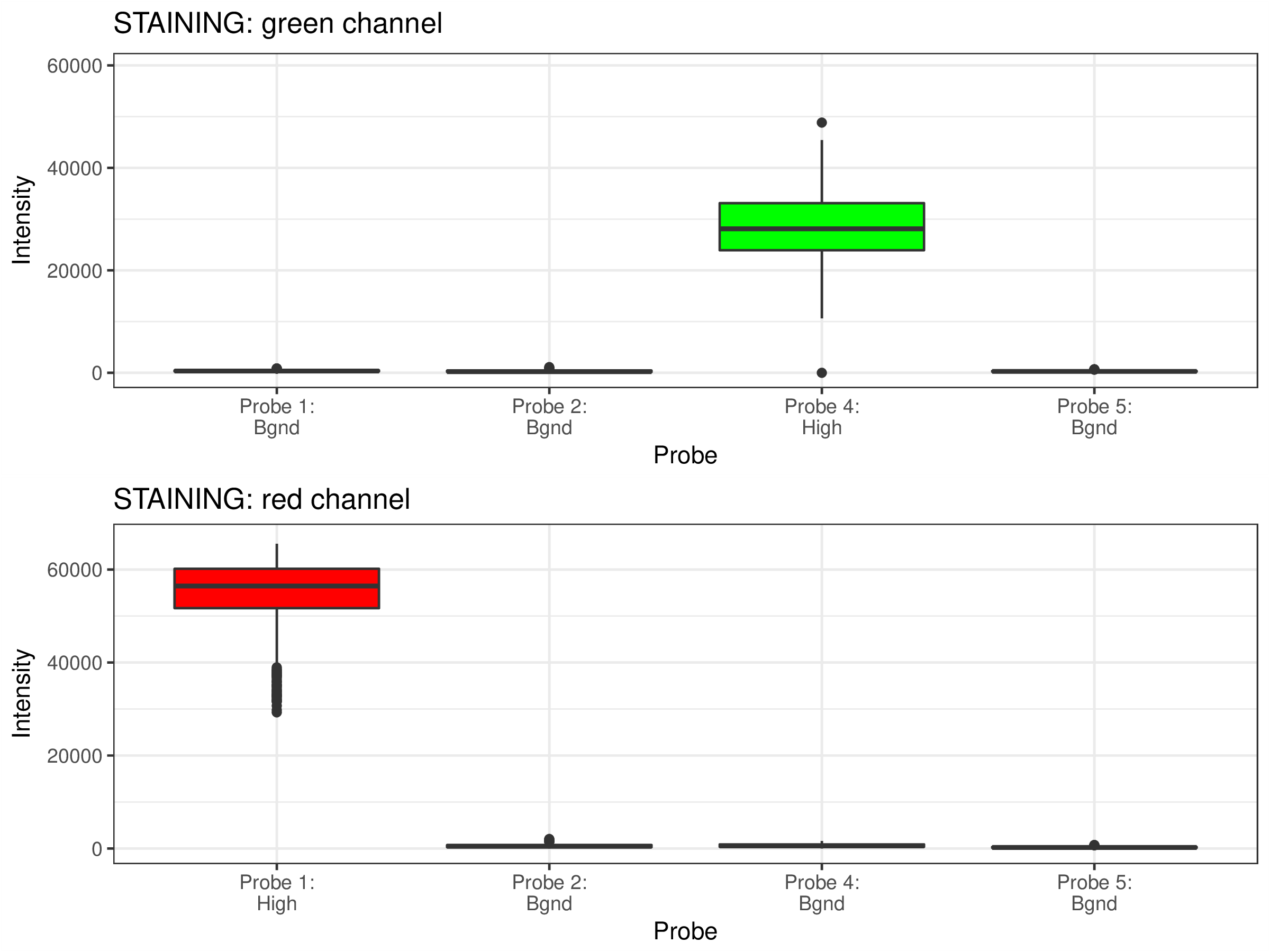
Quality Control Box Plots
Each box plot below shows the signal distribution of quality control probes across all samples. The control box plots are separated by control types. Detailed description of the control probes is given in the RnBeads vignette.
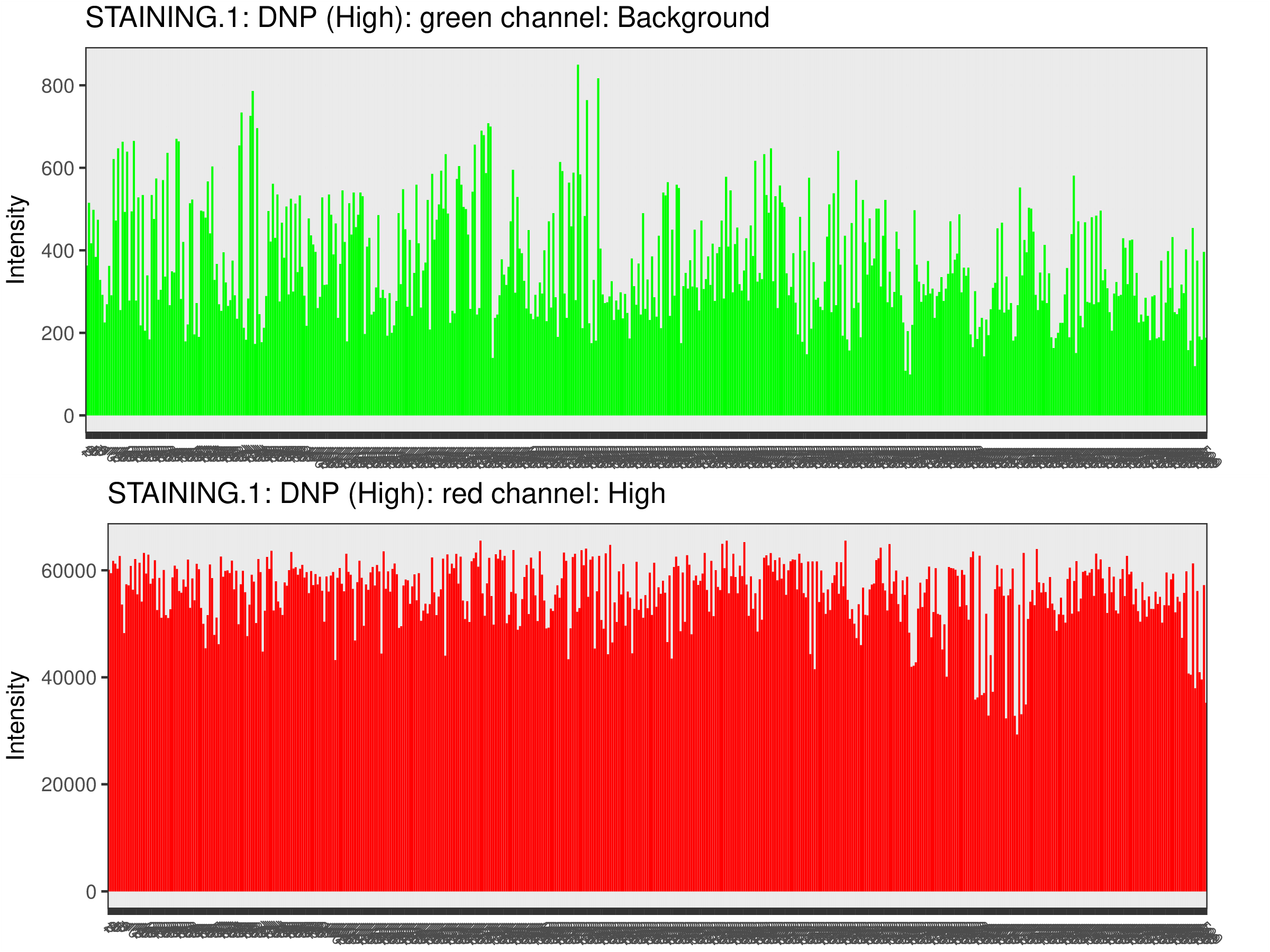
Quality Control Bar Plots
The plots below visualize the exact signal levels at each quality control probe. Note that the scale is not standardized. Background signal is usualy at the level of 1000 to 2000.
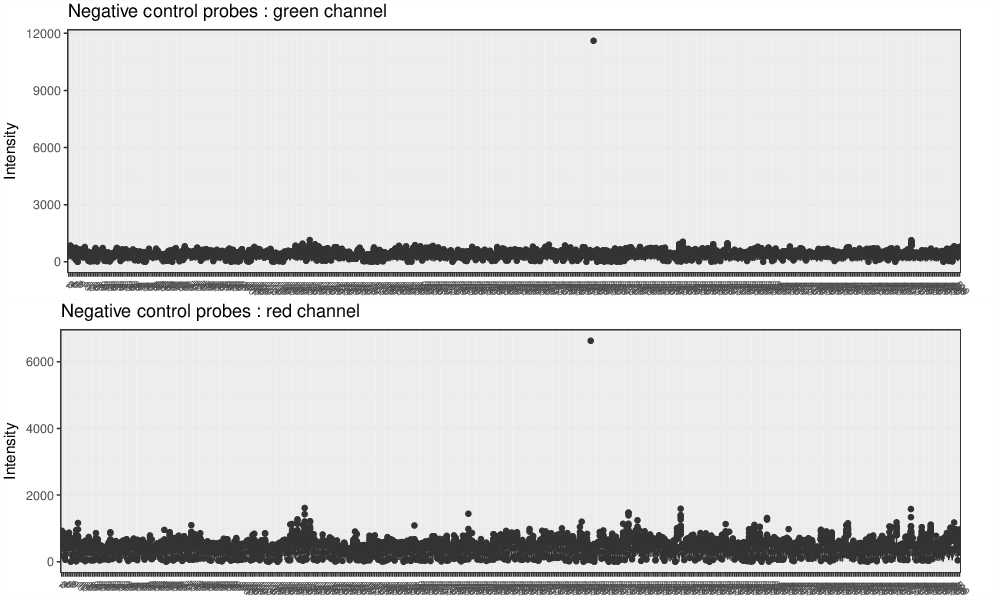
Negative Control Box Plots
Negative control box plots visualize background intensity distributions of all analyzed samples. Samples with skewed distributions and high medians are likely to be of low quality and should be discarded from the analysis.
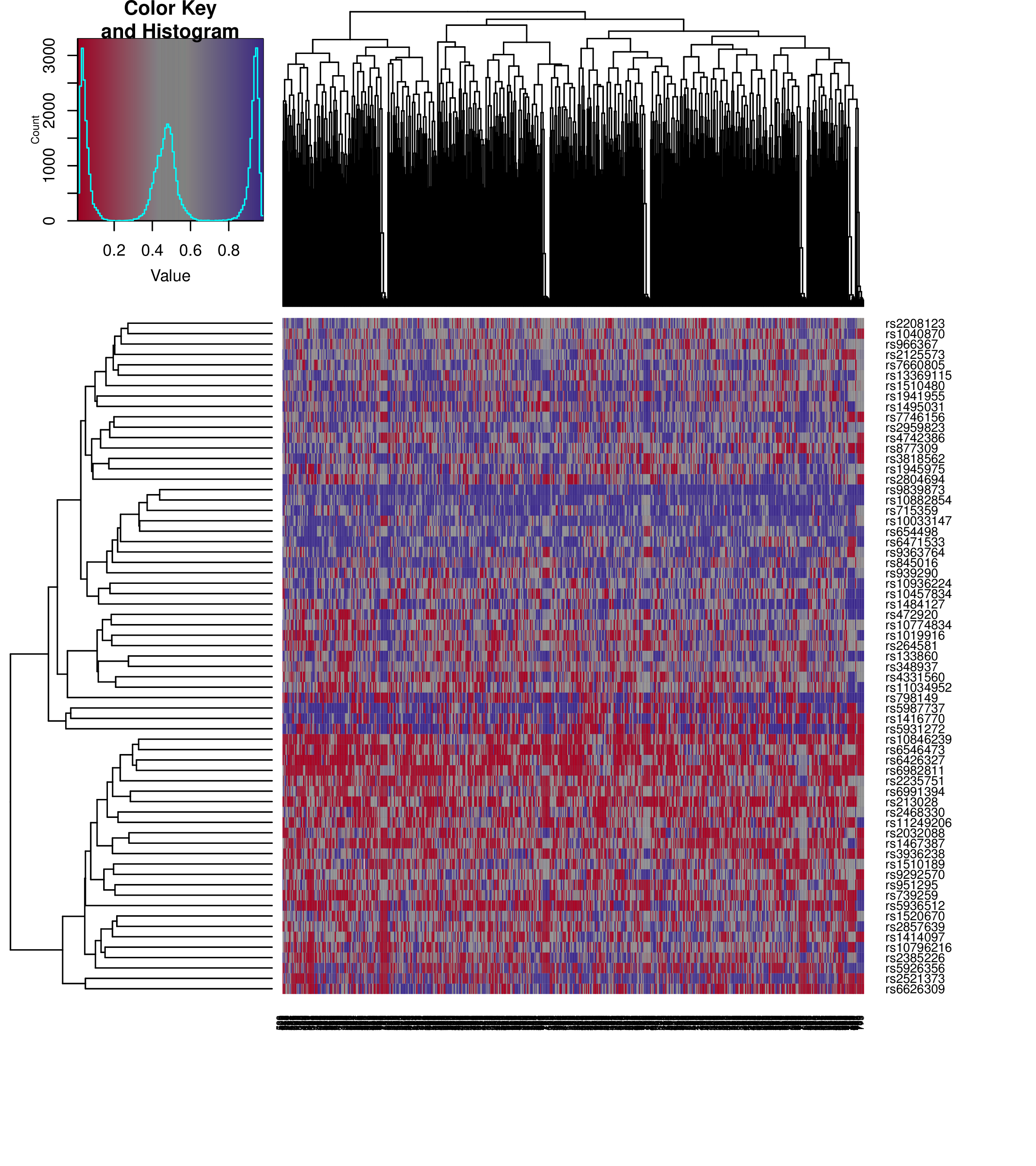
Analysis of the values of the SNP-based probes can help identify sample mixups.
SNP Heatmap
SNP heatmap enables the identification of sample mixups in particular for genetically matched designs.
SNP-based Distances
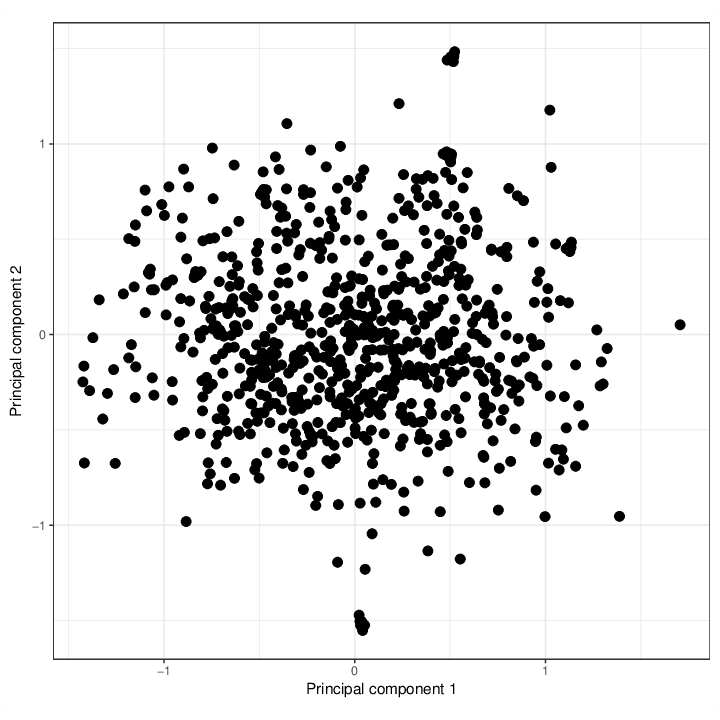
If we inspect the dataset in the space defined by the SNP probes only, samples appearing close to each other are genetically similar.
The figure below shows the samples in the first two principal components of the space defined by the 64 SNP probes.
The full table of all pairwise distances is stored in a dedicated comma-separated file accompanying this report.
Genetic Purity
The relative purity of a sample can be estimated by calculating the mean absolute distance from the observed β values of its SNP probes to an idealized profile, consisting of the values 0, 0.5 and 1 only. This score is referred to as genetic noise in this report. Samples with large genetic noise might contain subpopulations with genomic aberrations (deletions and amplifications), or be contaminated with DNA from multiple individuals. It is important to note that the genetic noise could be strongly influenced by batch effects and should therefore be considered only in the context of samples with similar profiles of intensity values.
The figure below lists the genetic noise of all samples in the dataset. The full table with these values is available in a dedicated comma-separated value file accompanying this report.
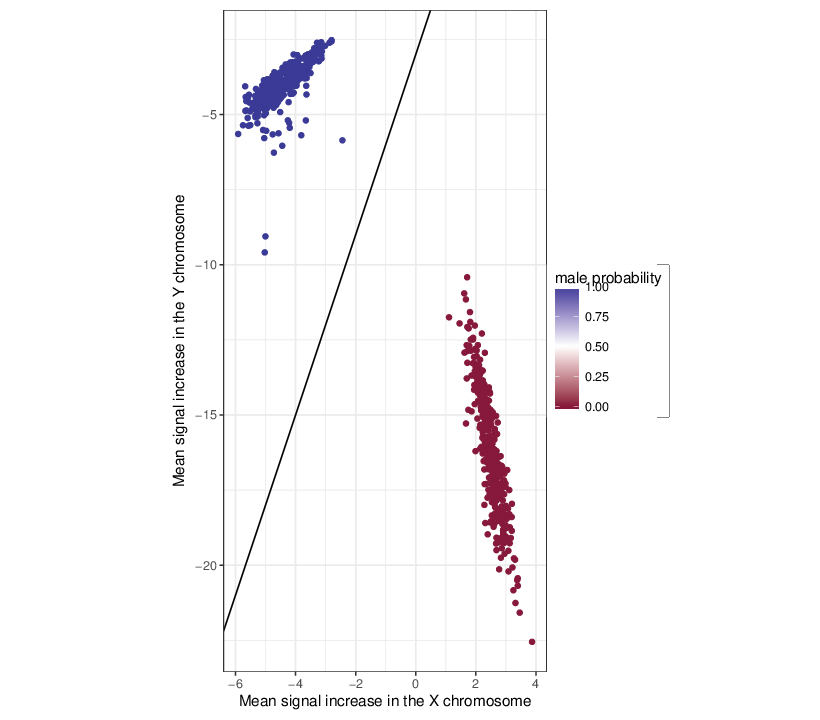
RnBeads predicted the gender of the samples in the dataset using a logistic regression model. The results are summarized in the table below.
| female |
390 |
| male |
402 |
| unknown |
0 |
Gender was predicted based on the increase (or decrease) of mean signal intensities in the sex chromosomes w.r.t. the corresponding value in autosomes. The figure below displays these characteristics of the samples.